Completely different mechanisms of adaptation have completely different timescales. Epigenetic adjustments are sometimes fast and reversible, whereas most genetic adjustments have almost negligible charges of reversion (1). This poses a problem for genetic adaptation to transient circumstances corresponding to drug therapy; mutations that confer drug resistance are sometimes deleterious within the absence of drug, and the second-site suppressor mutations are required to revive health (2, 3). Preexisting tandem repeats (satellite tv for pc DNA) bear frequent growth and contraction (4⇓–6). Whereas repeats are uncommon inside most coding sequences and purposeful components, there’s some proof for conserved repetitive areas that bear growth and contraction to control protein capabilities or expression (6⇓–8). RNA interference– or Chromatin-based epigenetic states have been related to transient drug resistance in fungi (9) and most cancers cells (10, 11), and transient resistant states have been characterised by variations in organelle state, progress price, and gene expression in budding yeast (12, 13). In micro organism and in fungi, copy-number acquire and subsequent loss can lead to reversible drug resistance (14⇓⇓⇓–18). Nonetheless, all genetic techniques developed to this point for learning unstable genotypes depend on reporter genes and thus examine just one genetic locus and just one sort of genetic change.
Unbiased, next-generation sequencing-based approaches might give a extra world view, permitting us to grasp the principles that govern unstable genotypes at a genome-wide scale. Nonetheless, genetic adjustments with excessive charges of reversion have a tendency to stay subclonal (19⇓–21), and it’s difficult to tell apart most varieties of low-frequency mutations from sequencing errors (22), particularly in advanced genomes with great amount of repetitive DNA or de novo duplicated genes. Thus, fast-growing organisms with comparatively small and easy genomes are significantly nicely fitted to figuring out whether or not transient mutations exist, for the genome-wide characterization of such mutations, and for identification of the underlying mechanisms.
Outcomes
Microhomology-Mediated Tandem Duplications in Particular Genes Brought on Reversible Phenotypes in Schizosaccharomyces pombe.
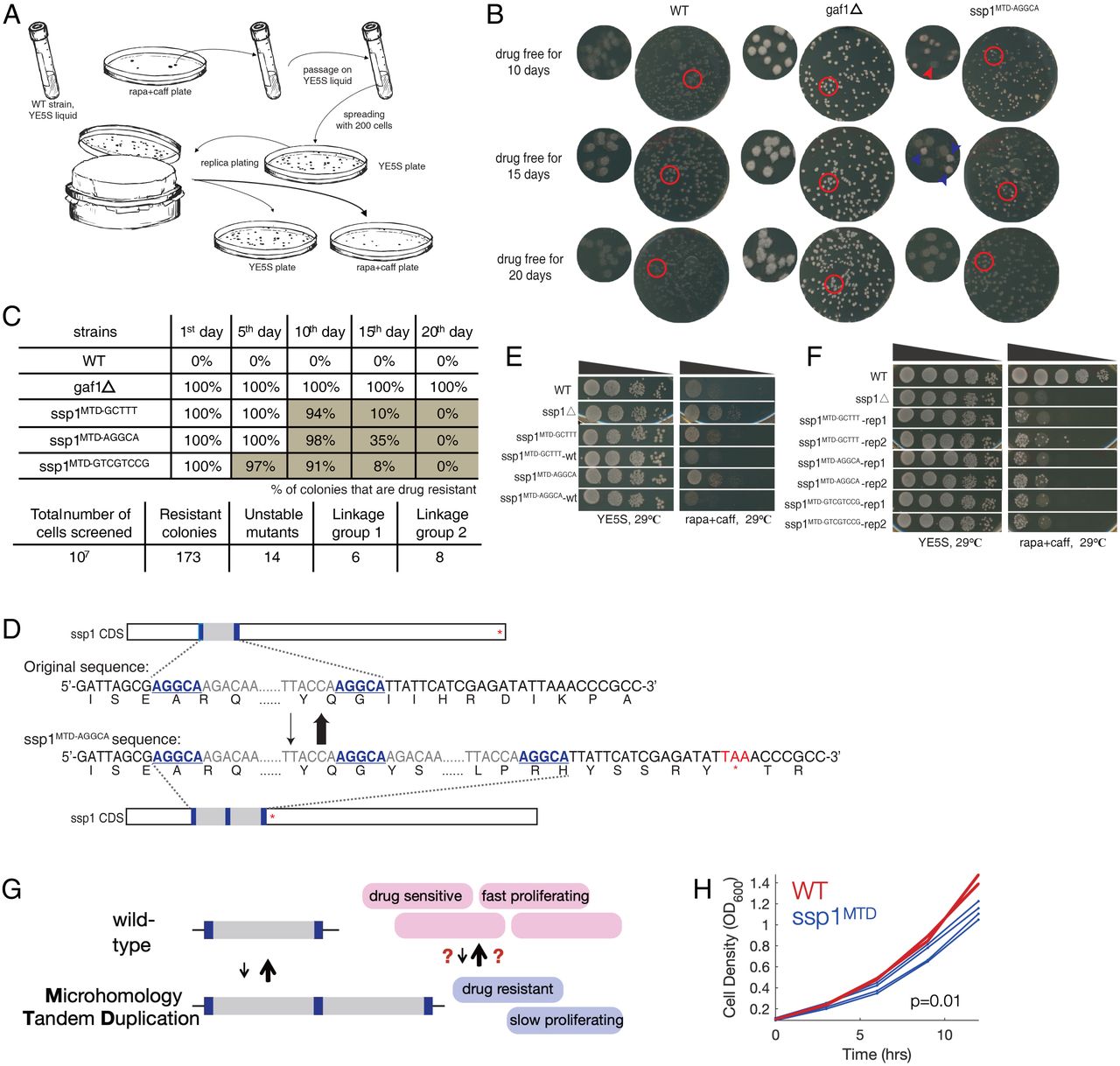
To find transient drug-resistance mechanisms in a eukaryote, we carried out a genetic display screen within the fission yeast Schizosaccharomyces pombe for spontaneous mutants which might be reversibly immune to rapamycin plus caffeine (caffeine is required for rapamycin to inhibit progress in S. pombe) (23) (Fig. 1A). We plated 107 cells from every of two unbiased wild-type strains to YE5S+rapamycin+caffeine plates and obtained 173 drug-resistant colonies, 14 (7%) of which exhibited reversible drug resistance following serial passage in no-drug media (Fig. 1 B and C). In distinction, resistance for deletion mutants corresponding to gaf1Δ (24) is irreversible, suggesting the existence of a sort of genetic or epigenetic alteration permitting for reversible drug resistance within the newly remoted strains (Fig. 1 B and C).

Display for mutants with unstable inherited resistance by rapamycin plus caffeine and establish extremely reversible mutations in ssp1. (A) Process to display screen mutants with unstable rapa+caff resistance utilizing delicate wild-type (WT) strains in S. pombe. (B) Unstable phenotype for one in all screened mutants on rapa+caff plates after reproduction plating. gaf1ρ as optimistic management exhibits sturdy and steady resistance. The times characterize for incubation time on drug free situation, permitting the expansion of resistance degenerated progeny. The purple arrows level to delicate progenies, whereas the blue to resistant ones. (C) Dynamics of reversion amongst recognized reversibly drug-resistant colonies. (D) Identification of tandem phase duplication in ssp1 for drug resistance progenies by whole-genome sequencing and reconfirmation by locus-specific PCR/Sanger sequencing. Underlined and daring bases stand for the MHP. The prematured cease codon is marked with purple. (E) ssp1 inactivation precipitated rapamycin resistance and the substitute of ssp1MTD sequence to wt-ssp1 rescue the drug resistance to WT stage. (F) Warmth -resistant isolates are ceaselessly obtained in ssp1MTD strains. (G) A cartoon of reversible MTDs that trigger drug resistance and a proliferation defect. (H) Development curves of WT (purple, two replicates) and ssp1MTDAGGCA (blue, 4 replicates).
We used genetic linkage mapping and whole-genome sequencing to establish the molecular foundation of reversible rapamycin+caffeine resistance. We recognized two linkage teams (SI Appendix, Fig. S1A); we couldn’t establish any frequent mutations within the first linkage group, suggesting an epigenetic or nonnuclear genetic mutation or an inheritable variation that continues to be to be detected. In distinction, all eight strains within the second linkage group contained tandem duplications within the gene ssp1, a Ca2+/calmodulin-dependent protein kinase (human ortholog: CAMKK1/2) which negatively regulates TORC1 signaling, the pathway inhibited by rapamycin, suggesting that mutations in ssp1 have been causal for drug resistance (25).
The ssp1 linkage group contained three insertion alleles, all of which have been tandem duplications of a brief DNA phase (55/68/92 base pairs [bp] in size) and had 5 to eight bp of an identical sequence (microhomology pairs, MHPs) at every finish (Fig. 1D and SI Appendix, Fig. S1B and Dataset 7). We postulate that these microhomology-mediated tandem duplications (MTDs) (26⇓–28) are essential for de novo technology of reversible mutations.
All three MTDs resulted in frameshifts and inactivation of ssp1. The same stage of drug resistance was discovered within the ssp1Δ, and substitute of the MTD alleles by transformation with wild-type ssp1 restored sensitivity (Fig. 1E). Sanger sequencing confirmed that every one 16 randomly chosen drug-sensitive revertants of the MTD alleles had the wild-type ssp1 sequence. Lastly, ssp1Δ and ssp1MTD strains are temperature delicate. Spontaneous drug-sensitive nonts revertants have been ceaselessly recovered for all of the ssp1MTD alleles at a frequency of roughly 1/10,000 cells however not for the ssp1 deletion (Fig. 1F). The frequency of revertants is thus 100× increased than the ahead MTD frequency (8/107), and MTDs in ssp1 are causal for reversible temperature sensitivity and drug resistance.
Supporting the notion that MTDs will not be particular to rapamycin/caffeine therapy and/or the goal gene ssp1, in an unrelated genetic display screen for suppressors of the gradual progress defect of cnp1-H100M, a degree mutation within the centromere-specific histone gene, we recognized MTDs within the transcription repressor genes yox1 and lsk1 (SI Appendix, Figs. S1B and S2 and Dataset 7). These MTDs improve health within the cnp1-H100M background, and subsequently, not like ssp1MTDs, revertants don’t improve in abundance within the mutant background. Nonetheless, within the ssp1wt background, these MTDs are deleterious, and revertants accumulate (SI Appendix, Figs. S1 and S2). Thus, MTDs should not gene particular and certain happen all through the genome.
10,000× Complete-Genome Sequencing Recognized Hundreds of Subclonal MTDs inside a Clonal Inhabitants.
Based mostly on the size of the preliminary genetic display screen and assuming drug resistance isn’t induced by rapamycin, the frequency of cells with any protein-inactivating MTD in ssp1 in an exponentially rising nonselected wild-type inhabitants is estimated ∼8 × 10−5. This end result suggests {that a} clonal, presumed “isogenic” inhabitants comprises all kinds of subclonal MTDs at a number of loci all through the genome. The frequency of any single MTD will depend upon the speed of MTD formation, the speed of reversion, and the health price of the MTD (19⇓–21). The health defect imposed by the MTD might be on account of altered gene expression or protein operate or from the health price of ∼0.025% per kb of extra DNA (29, 30).
To establish the cis-encoded determinants of MTD frequency, we developed a computational pipeline for detecting subclonal MTDs in high-coverage Illumina sequencing information (see Supplies and Strategies for particulars). This methodology first identifies all MHPs in a DNA phase or genome and generates “signatures” for sequences that may be created by every attainable MTD. It then identifies sequencing reads that match these signatures and thus offers experimental assist for the existence of a specific MTD inside the inhabitants (Fig. 2A). This methodology is able to figuring out subclonal MTDs current at very low frequencies within the inhabitants.

Identification and verification of subclonal MTDs from ultra-deep sequencing information. (A) The computational pipeline finds all sequencing reads whose ends don’t match the reference genome and checks whether or not the reads as a substitute match the sequence that may exist on account of an MTD. Proven are reads recognized within the pipeline, aligned to both the reference genome (Prime) or to an artificial genome with the MTD (Backside). Crimson and blue mark reads that map to reverse strands are proven. The MHPairs are proven in darkish blue, and positions in every learn that don’t match the reference are coloured in accordance with the bottom within the learn. (B) The common frequency of sequencing reads that assist every MTD in ssp1 from 106 protection sequencing of the gene from S. pombe, from a plasmid-borne ssp1 in E. coli, or from a chemically synthesized fragment of the ssp1 gene. Error bars are SEM throughout replicates. (C) The genomic places of 314 MTDs that happen in just one single MA line and reanalyzed uncooked sequencing information from ref. 32; error bars are SD from bootstrapping. The Prime row exhibits the % of MTDs present in important genes, in genes whose haploid deletion mutant is viable however exhibits a health defect (33), in all nonessential genes, or in intergenic areas, individually for haploid and diploid MA strains. The Backside row exhibits the % of MTDs in every class that create a tandem duplication that has a size divisible by three and, for intragenic MTDs, don’t disrupt the studying body. The purple dashed line exhibits the random expectation (one-third of MTDs).
To find out whether or not subclonal MTDs captured by sequencing characterize the true genetic variation or are technical artifacts (31), we carried out two orthogonal assessments. Within the first, we examined whether or not MTDs are particular to genomic DNA or additionally exist in chemically synthesized DNA. We carried out 105x − 106x protection sequencing of ssp1 DNA fragments PCR-amplified from genomic DNA from a cloned copy of the gene in a plasmid in Escherichia coli or chemically synthesized 150-nt and 500-nt fragments of the gene in addition to direct sequencing of chemically synthesized quick DNA fragment and plasmid-borne fragment with out PCR amplification. We noticed way more MTDs within the pombe genomic DNA than within the chemically synthesized or plasmid borne controls (Fig. 2B and SI Appendix, Fig. S3), suggesting that MTDs are largely not brought on by PCR or an artifact of Illumina sequencing. It’s unclear why the plasmid-borne copy of ssp1 has fewer MTDs than the chemically synthesized DNA, but it surely raises the chance that MTDs could also be extra frequent in eukaryotes (Fig. 2B and SI Appendix, Figs. S6–S8). We detect MTDs within the E. coli genome 1/twentieth as usually as in S. pombe and 1/sixtieth as usually as in Saccharomyces cerevisiae (SI Appendix, Fig. S9).
As a second check, we hypothesized that the majority MTDs in important genes ought to be deleterious and recessive. We subsequently analyzed uncooked sequencing information from 209 S. cerevisiae haploid and diploid mutation accumulation (MA) strains (32) and recognized all MTDs that happen in just one MA line. In haploids, MTDs have been depleted in each important genes and nonessential genes whose deletion causes a health defect (Fig. 2C). As well as, the MTDs that did happen have been extra more likely to preserve the right studying body; the only MTD in an important gene in a haploid was subclonal, maintained the right studying body, and was simply 112 bp from the three′ finish of CCT7, a 1,652-bp gene. In distinction, there was no such reading-frame bias in diploids, nonessential genes, or intergenic areas (Fig. 2C). Subsequently, uncommon subclonal MTDs recognized by ultra-deep sequencing are possible actual organic occasions largely not experimental artifacts.
To evaluate the prevalence of MTDs and to establish the sequence-based guidelines that decide the likelihood of formation of every tandem duplication, we grew a single diploid fission yeast cell as much as ∼108 cells (25 generations) and carried out whole-genome sequencing to a median protection of 10,000× the diploidy relaxed choice, permitting mutations to build up all through the S. pombe genome.
With 10,000× genome sequencing, we recognized 5,968 (0.02%) MHPs by which a number of sequencing reads supported an MTD. We noticed zero MTDs in most genes, possible on account of under-sampling (SI Appendix, Fig. S4). Nonetheless, 20 genes contained greater than 10 completely different MTDs in a single “clonal” inhabitants (Fig. 3A). To know this heterogeneity throughout the genome, we used a logistic regression machine-learning mannequin to foretell the likelihood of duplication at every MHP. MH size, guanine and cytosine (GC) content material, inter-MH distance, measured nucleosome occupancy, transcription stage, and an area clustering on the size of 100 nt have been capable of predict which MHPs give rise to duplications with an space beneath the curve rating of 0.9 with 10-fold cross validation (Fig. 3 B and C and SI Appendix, Fig. S5 and Dataset 5). We be aware that the height at 150-nt inter-MH spacing is unbiased of learn size, was not present in E. coli or in mitochondrial DNA, and varies between haploid and diploid (SI Appendix, Figs. S5–S8). This evaluation revealed that properties of MHPs considerably have an effect on the probability of MTD formation; for instance, and in step with earlier work in E. coli (34), lengthy GC-rich MHP is 1,000× extra more likely to generate a tandem duplication than a brief AT-rich one.

Identification of the cis-determinants of MTD by means of ultra-deep sequencing and identification of subclonal duplications. (A) A histogram of the variety of MTDs present in every gene from 10,000× whole-genome sequencing. (B) The 25 million MHPs within the genome have been binned in teams of 10,000 with the identical MH sequence size and comparable GC content material (Left) or inter-MHPair distance (Proper) and the share of MHPs in every group with an noticed MTD was calculated. A logistic regression mannequin was educated with 10-fold cross-validation to foretell the likelihood of observing an MTD at every MHPair. (C) The space from every MHP to the closest MHP with an MTD was calculated, and the share of MHPs with an MTD was calculated for MHPs lower than (purple) or farther than (inexperienced) 100 nt from the closest MHP. (D) For every 1-kb window within the genome, proven are the variety of MHPairs (purple), the variety of noticed MTDs (blue), and the anticipated variety of MTDs from the logistic regression mannequin (inexperienced). (E) An instance chilly spot (0.2MTDs/kb) and scorching spot (0.7 MTDs/kb) in chromosome I. The chilly spot has fewer MTDs after considering the variety of MHPs, (Fisher’s precise check, P = 2.76 × 10−9, odds ratio = 3.843). (F) The sum of scores from the logistic regression mannequin for every MHP in every gene, with the genes grouped by the noticed variety of MTDs within the 10,000× protection information.
Whereas MHPs are unfold roughly uniformly all through the genome (Fig. 3D, purple), we noticed each scorching spots, by which MH-mediated technology of tandem duplications are frequent, and chilly spots, by which they’re uncommon (Fig. 3E). Native variations in MHP density can solely clarify a few of the hotspots, whereas our logistic regression mannequin explains the overwhelming majority, suggesting that hotspots with frequent formation of tandem duplications are largely decided by the native DNA sequence options along with microhomologies. The consequence is that duplications are greater than 10× extra more likely to happen in some genes than others, and this variation is appropriately predicted by our mannequin (Fig. 3F). We detected no MTDs in ura4, which has a rating of 52, inserting it within the backside third of genes (SI Appendix, Fig. S10 and Dataset 4) and offering a attainable reason why MTDs haven’t been seen in 5-FOA-based screens of mutations in ura4 (35). Our outcomes additionally emphasize that high-coverage sequencing is critical to establish ample numbers of MTDs; 1 billion reads could be required to establish half of the 25 million attainable MTDs within the S. pombe genome (SI Appendix, Fig. S4).
We recognized three completely different subclonal MTDs within the SAGA advanced histone acetyltransferase catalytic subunit gcn5, inserting gcn5 within the prime 5% of genes for each noticed and predicted MTDs, suggesting that MTDs in gcn5 ought to be discovered ceaselessly in a genetic display screen. Certainly, examination of 16 beforehand recognized (36) suppressors of htb1G52D recognized MTDs in gcn5, in addition to in ubp8, by which we additionally noticed an MTD in our high-coverage sequencing information (SI Appendix, Fig. S1B). These outcomes counsel that MTDs come up in most genes at a high-enough frequency inside populations to be able to be the uncooked materials on which pure choice acts.
Replication Slippage Modulates the Charge of MTD Reversion at ssp1.
Having established that native cis-encoded options decide the frequency with which tandem duplications come up from MHPs, we subsequent sought to establish the trans-genes that have an effect on the MTD course of. ssp1MTD alleles fail to develop at 36 °C, and their reversion again to wild sort suppresses the temperature sensitivity, offering a approach to measure the consequences of mutations on reversion frequency. We screened a panel of 364 strains with mutations in DNA replication, restore, recombination, or chromatin group genes for mutants that have an effect on the speed of ssp1MTD reversion again to wild sort (Dataset 6) and located three mutants that considerably elevated and eight that considerably decreased the frequency of ssp1WT revertants (Fig. 4 A–C).

A genetic display screen to establish the regulators of MTD reversion. (A and B) Surveyed mutants confirmed diminished ssp1MTD reversion frequency represented by TS restoration phenotype. The non-TS phenotype of single mutation and ssp1ρ alone or mixed with different mutants retained extreme temperature-sensitive phenotype at 36 °C ought to be established. The variety of TS revertants beneath 36 °C point out the reversion frequency of ssp1MTD. The preliminary gradient for recognizing assay was 105 cells and diluted with 10-fold gradient (cell quantity: 105, 104, 103, 102, and 101). (C). Quantification of ssp1MTD reversion frequency in mutants (n ≥ 3 organic repeats, error bars are s.e.m., ***P < 0.001, **P < 0.01, and *P < 0.05 t check in comparison with wt). (D) Two colonies of WT and two of every mutant have been picked and SPCC1235.01 amplified by PCR and sequenced to 106 protection. Present is the typical throughout the 2 replicates of the MTD frequency at every of the three,002 MHPs. (E) The share of MHPairs with a number of reads in assist of an MTD in SPCC1235.01. (F) For all MHPairs with an MTD, the frequency of reads supporting that MTD per 106 reads that map to that MHPair.
Replication fork collapse is a serious supply of double-stranded breaks (DSBs), and the following homologous recombination (HR)–associated restarting course of is error inclined and is understood to generate microhomology-flanked insertions and deletions by way of replication slippage, a course of by which, when replication resumes for a stalled or collapsed fork, the unwound nascent strand could anneal with a homologous phase on the template, both on the neighborhood (37, 38) or at a distance (34) of the paused website, with ensuing replication on noncontinuous template. Inactivation of Rad50, Rad52, or Ctp1 leads to decreased replication slippage and decreased MTD reversion (37, 39) (Fig. 4 A–C). Deletions of mhf1 and mhf2, two subunits of the FANCM–MHF advanced, which is concerned within the stabilization and transforming of blocked replication forks, additionally decreased the frequency of MTD revertants. It’s subsequently possible that replication slippage throughout HR-mediated fork restoration causes reversion of MTDs in these mutants and could possibly be one contributing think about wild-type background.
Replication stresses activate a checkpoint that promotes DNA restore and restoration of stalled or collapsed replication forks and delays entry into mitosis (40, 41). The inactivation of replication checkpoint kinase cds1 or its regulator mrc1 could thus lead to a failure to revive the replication fork, inflicting elevated genome instability and MTD reversion. The replication checkpoint would thus be required for the steadiness of MTDs. Persistently, we discovered that deletion of the DNA harm checkpoint kinase cds1 or its regulator mrc1 elevated the frequency of ssp1WT revertants. Deletion of the single-stranded DNA binding A (RPA) subunit ssb3 (RPA3/RFA3) or the multifunctional 5′-flap endonuclease rad2 additionally elevated the frequency of revertants (Fig. 4C).
Many genes recognized within the display screen are multifunctional and play roles in each replication and restore. We subsequently carried out quantitative epistasis evaluation to find out the relation between six of the recognized genes and the Mediator of the Replication Checkpoint, mrc1, which interacts with and stabilizes Pol2 at stalled replication forks. Along with the checkpoint activator cds1, deletion of rad2 had no impact in an mrc1Δ background, suggesting that every one three of those genes act in the identical pathway (Fig. 4D). In distinction, deletion of ssb3 elevated the frequency of revertants in each wild-type and mrc1Δ backgrounds, and deletion of pds5 or rik1 decreased the frequency of revertants in each wild-type and mrc1Δ backgrounds, although to not the extent anticipated for genetic independence, suggesting partial epistasis. In distinction, the consequences of rad50 deletion have been fully unbiased of mrc1 (Fig. 4D).
Whereas the noticed numbers of MTDs in ultra-deep sequencing experiments are a operate of each duplication and reversion charges and the entire above genes could play a task in each processes, the above outcomes steered that on account of elevated reversion charges, the quantity and frequency of MTDs could be diminished in cds1Δ and rad2Δ strains. To check this concept, we carried out 106x protection sequencing of the hotspot gene SPCC1235.01. We observe MTDs at fewer MHPs and an general lower within the variety of MTDs in each mutants (Fig. 4 E and F).
Half of Insertions and Tandem Duplications in Pure Isolates Are MH Mediated.
It was baffling that MTDs are prevalent inside populations and that the primary theoretical proposal for microhomology-mediated processes within the technology of tandem duplications is 20 y previous (5); but, comparatively little is understood in regards to the ahead course of and even much less in regards to the reversion, suggesting that these occasions should not usually encountered or recognized as such. To higher perceive the dynamics of MTDs inside a inhabitants, we used a easy mannequin of impartial mutations inside a rising inhabitants that takes under consideration each ahead and reverse mutation charges and commenced with 100% of people as wild sort (see Supplies and Strategies). The mutant frequency at all times will increase and over quick timescales (Fig. 5 A, Left), rising the reverse price from being equal to the ahead mutation price (grey) to being 10,000 instances increased (yellow) has little impact.

MTDs stay subclonal due to excessive reversion charges, but half of insertions and de novo tandem duplications in pure populations come up at microhomology sequence pairs. (A) Simulations displaying the frequency of a impartial mutation (ahead mutation price = 10−7) inside a rising inhabitants at three completely different reversion charges (colours). Left and Proper present the identical simulates at completely different timescales, with the impact of reversion solely obvious at lengthy timescales. (B) A cartoon displaying three attainable varieties of microhomlogy-mediated insertions: easy insertion, tandem duplication, and better copy repeat. (C) Quantification of all insertions of at the very least 10 bp fastened in any of the 57 pure S. pombe isolates that characterize many of the genetic range inside the species, relative to the reference genome. Insertions have been categorized in accordance with the presence (purple) or absence (inexperienced) of actual MHPs on both aspect of the insert and to the kind of insert. There are 113 MTDs in wild pombe strains (second column). The suitable-most column (>1× -> >N×) refers back to the growth of repeats current within the reference genome. (D) Distributions of the anticipated MTD rating from the logistic regression mannequin (Left) and the variety of experimentally noticed subclonal MTDs (Proper) for genes with a number of microhomology-mediated insertions (purple) or for genes with no MH-mediated insertions (inexperienced) in any of the pure isolates. P values are from a Mann–Whitney U check.
Over longer timescales, excessive reversion charges trigger the mutant frequency to plateau and stay subclonal (Fig. 5 A, Proper), decreasing the fraction of impartial MTDs inside a inhabitants. Nonetheless, regardless of the excessive reversion price, each drift and choice allow fixation of MTDs inside a inhabitants. To establish fastened microhomology-mediated insertions, we searched the genome sequences of 57 wild S. pombe isolates (42) and located that fifty% of insertions bigger than 10 bp contain microhomology repeats (Fig. 5 B and C). Amongst these have been 158 microhomology-mediated insertions that didn’t include an apparent duplication and 113 MTDs with an MTD.
To check whether or not the propensity of MTD formation inside the laboratory pressure is predictive of extant sequence variation noticed in pure isolates, we examined whether or not the MTD rating for every gene predicts the probability of microhomology-mediated insertions in that gene. We discovered that genes with microhomology-mediated insertions in pure isolates are inclined to have increased predicted MTD scores and extra experimentally noticed MTDs (Fig. 5D), suggesting that the native options that have an effect on MTD formation within the laboratory additionally form evolution in nature.
MHPs with Longer MH Sequences Are Extra More likely to Generate MTDs that Preserve the Appropriate Studying Body.
We discovered that MHPs with longer MH sequences usually tend to kind MTDs. If the excessive propensity to generate MTDs has formed the S. pombe genome, any signature of choice ought to be stronger at MHPs with longer MH sequences and also needs to be stronger in important genes versus nonessential genes. We subsequently divided the 25 million MHPs with an MH size of 4 to 25 nt and an inter-MH distance of three to 500 nt into these totally contained inside in intergenic areas or totally contained inside important or nonessential genes and cut up them by MH sequence size. Particularly in coding sequences, MHPs at which an MTD wouldn’t disrupt the studying body are extra frequent than anticipated by probability, and this enrichment is increased in important genes and at longer MH sequences (Fig. 6). On the identical time, MHPs inside genes are extra frequent that anticipated by probability (SI Appendix, Fig. S12). Subsequently, pure choice has acted to lower the variety of MHPs that may create probably deleterious MTDs, and this choice is weaker for MHPs that may create an in-frame MTD.

Body-shifting MHPs are depleted from important genes in an MH sequence size–dependent method. (A) The variety of MHPs within the S. pombe genome with completely different MH sequence lengths (colours) for which an MTD would generate various insert sizes (x-axis). X-axis grid strains mark MTDs with insertion sizes divisible by three. Left exhibits MHPs which might be intergenic and Proper MHPs which might be totally contained with a coding sequence of a gene. (B) The share of MHPs with lengths evenly divisible by three (y-axis) for every MH sequence size (x-axis) which might be present in intergenic areas (blue), totally contained inside important genes (black) or inside nonessential genes (purple). Random expectation is that one-third of MHPs could have an insert measurement evenly divisible by three (orange).
Taken collectively, our outcomes exhibit that MTDs happen ceaselessly and broadly all through the genome inside a clonal inhabitants. These findings point out that prime ranges of subclonal genetic divergence are prevalent however are beneath detected utilizing typical sequencing approaches that are inclined to disfavor the detection of low-abundance subclonal variants. As many MTDs create massive insertions, they’re extra more likely to be deleterious. Nonetheless, MTDs present plasticity to the genome and its performance, for instance, by permitting cells to grow to be drug resistant, whereas permitting the resistant cell lineage to revert again to wild sort and regain excessive health as soon as the drug is eliminated. Choice can act on this genetic range for its reversibility or by utilizing the tandem duplications because the preliminary step for the technology of upper copy quantity repeats, that are evolutionarily fastened in extant genomes and historically thought to be a serious supply of genome divergence. Whereas earlier work has proven that preexisting repeats bear quickly reversible adjustments, the sequence-encoded guidelines regulating the beginning and demise of such sequences have been much less studied (34). This work reveals that quite a few websites all through the genome have the potential of evolving into such repetitive components. Moreover, MH sequence size–dependent depletion of frame-shifting MHPs in important genes exhibits that pure choice has formed the genome to keep away from MHPs that may ceaselessly generate deleterious MTDs. Lastly, a lot in the identical approach as repetitive DNA could have been positively chosen for as a regulatory aspect to keep up reversible genetic range (7, 8), the big variety of MHPs that may lead to in-frame MTDs raises the chance that some genes could preserve MHPs to technology purposeful genetic range, making a dynamic protein-coding genome.
Dialogue
Why haven’t MTDs been recognized extra ceaselessly in genetic screens and MA assays? There are a number of attainable causes. Mutation callers are ineffective in detecting lengthy insertions from quick reads: on simulated information, each Mutect2 and HaplotypeCaller usually fail to detect tandem duplications longer than 85 bp. Additionally, MTDs are sometimes recognized as insertions however not particularly as MTDs (SI Appendix, Figs. S1B and S11), suggesting the necessity for computational instruments for figuring out MTDs. URA4 and URA3 have comparatively few MHPs (SI Appendix, Fig. S10). In lots of 5-FOA–primarily based mutation spectra papers, solely substitutions have been analyzed intimately, as indels don’t happen with high-enough frequency to generate good statistics (43). As a result of excessive price of reversion, it’s possible that tandem duplications in URA4 would yield URA+ colonies when restruck onto -URA plates; this excessive reversion price could result in MTD-containing colonies being discarded in lots of various kinds of genetic screens. In our reanalysis of S. cerevisiae MA strains, nearly all de novo MTDs have been subclonal (SI Appendix, Fig. S11B). With new computational instruments for figuring out MTDs plus third-generation sequencing platforms with improved potential to detect lengthy indels, it’s possible that MTDs might be implicated in additional phenotypes.
Unbiased genome-scale approaches have been very informative for the mechanisms that generate level mutations in each wild-type and mutant cells (44). It’s clear that a number of molecular mechanisms can provide rise to tandem duplications in microhomology-dependent and unbiased manners, and the mechanisms could differ in mutants and between species. In plant mitochondria genomes, longer MH sequences are related to longer tandem duplications, suggesting that microhomology is concerned within the technology of tandem duplications, possible by way of microhomology-mediated repairing of DSBs (45, 46) or slippage strand replication (47). In distinction, tandem duplications within the rice nuclear genome are inclined to don’t have any or shorter microhomology sequences, suggesting that within the rice nuclear genome, tandem duplications possible kind by way of patch-mediated DSB creation adopted by NHEJ (48). In E. coli, the lagging-strand processing exercise of Pol I is required for stress-induced, MH-mediated amplification of 7- to 32-kb segments (34). Nonetheless, easy fashions can’t account for all options noticed throughout research, and it’s clear that a number of mechanisms play a task (49⇓–51).
All present strategies of measuring microhomology-mediated duplications and deletions impose synthetic size scales, whereas genetic screens require your entire gene or a particular area be duplicated. Thus, whereas microhomologies are related to deletions on the 500-bp to 1-kb vary in E. coli (52) and with unstable amplifications of seven to 37 kb (16) and choice for elevated gene expression usually enriches for ∼12-bp MH-mediated amplification of ∼10kb (53), these size scales are decided by the places of MH sequences within the specific genomic area required to be duplicated within the genetic display screen and the scale of the area required to be duplicated or amplified.
Genome-wide sequencing-based approaches are much less biased however nonetheless not bias free. We restrict the length-scale to 500 bp and set decrease and higher bounds of three bp and 25 bp for the MH sequences. Whereas the MTD frequency relative to the variety of 1-bp or 2-bp MH sequence pairs within the genome is more likely to be low, the variety of 2-bp and 1-bp MH sequences is excessive. Preferential flanking of tandem duplications by 2-bp and even 1-bp sequence identities have been reported (27, 28, 45, 54), suggesting that subclonal MTDs generated by quick MH sequences could also be frequent. Whereas it’s computationally intractable to use the directed “signatures” methodology offered right here to look MH pairs separated by greater than 500 bp, this methodology could possibly be prolonged to >500 bp if the search is restricted to longer MH sequences, that are uncommon however drive excessive charges of MTD formation (28, 34). Equally, the strategy could possibly be prolonged to damaged microhomologies (55) however with a fair increased computational price. Computational approaches utilizing third-generation (Nanopore and PacBio) sequencing have the potential to supply a really unbiased measure of duplication and deletion frequencies in addition to reply questions on how usually amplifications are extrachromosomal versus intrachromosomal (17, 56) and tandem versus inverted amplifications (57). Verification of those algorithms will take some work, as Nanopore and Pac-Bio generally give completely different outcomes when sequencing tandem repeats (58).
As a result of completely different molecular mechanisms have completely different sequence necessities, replication strand biases, and size scales (34, 50), genome-wide unbiased strategies are mandatory to grasp the relative contribution of every mechanism to MTD formation and collapse. As they will, in concept, measure occasions throughout any distance, from single base pair to interchromosomal, and at a limiteless variety of completely different loci, with variation in chromatin contexts, transcription, and different genome-architecture options, ultra-deep sequencing is probably going one of the simplest ways to quantitatively perceive the varied organic mechanisms that contribute to the dynamic genome.
Comments
0 comments